 Blog HemoClass
Blog HemoClass
png.jpg)
Linfócito reativo em crianças: por que é um achado normal (e como interpretar sem pânico)
Warning: strtotime(): It is not safe to rely on the system's timezone settings. You are *required* to use the date.timezone setting or the date_default_timezone_set() function. In case you used any of those methods and you are still getting this warning, you most likely misspelled the timezone identifier. We selected the timezone 'UTC' for now, but please set date.timezone to select your timezone. in /home/hemocla/public_html/site/blog.php on line 179
Warning: date(): It is not safe to rely on the system's timezone settings. You are *required* to use the date.timezone setting or the date_default_timezone_set() function. In case you used any of those methods and you are still getting this warning, you most likely misspelled the timezone identifier. We selected the timezone 'UTC' for now, but please set date.timezone to select your timezone. in /home/hemocla/public_html/site/blog.php on line 179
21/10/2025 |
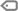 #Leucograma
#Padronização
#Hematoscopia
#exames
#Leucograma
#Padronização
#Hematoscopia
#exames
O que é um linfócito reativo?
É um linfócito que “ganhou corpo” após ativação antígeno–anticorpo. Essa ativação leva a alterações morfológicas (citoplasma mais amplo e basofílico, contorno irregular, às vezes nucléolo evidente) e funcionais (produção de anticorpos, citocinas etc.). Em outras palavras: é o linfócito fazendo o seu trabalho.
Por que aparecem tanto em crianças?
Exposição constante a antígenos novos: cada vacina, infecção banal e contato ambiental apresenta “novos cartões de visita” ao sistema imune.
Maturação imunológica acelerada: nos primeiros anos, há uma verdadeira “academia” para linfócitos B e T.
Janela etária típica: do nascimento até ~3,5–4 anos, é comum ver LR no esfregaço.
Resumo prático: em pediatria, encontrar LR é normal e esperado. Um hemograma de uma criança de ~2 anos completamente “estéreo” de LR pode, inclusive, levantar sobrancelhas — sempre dentro do contexto clínico.
Como interpretar no hemograma
1) Não olhar só para a contagem total
LR podem aparecer com leucócitos normais. “Só libero LR quando há leucocitose” é um erro. Contexto é rei: quadro clínico, diferencial leucocitário, morfologia.
2) Valor absoluto vs. percentual
Relate percentual e, quando possível, o valor absoluto. Pequenos percentuais em amostras com linfocitose típica da infância ainda são condizentes com fisiologia.
3) Descrição morfológica ajuda o clínico
Se o laboratório utiliza microscopia, vale caracterizar brevemente (p. ex.: “linfócitos com citoplasma basofílico amplo, contorno irregular, compatíveis com reatividade”). Evite termos alarmistas.
Quando o LR não é só fisiologia? (sinais de alerta)
Embora raro, há contextos que pedem atenção e correlação clínica:
Citopenias associadas (anemia, trombocitopenia relevantes)
Aparência atípica não compatível com reatividade usual (suspeita de blastos)
Sintomas sistêmicos importantes (perda ponderal, febre prolongada, sangramentos, linfonodomegalias exuberantes)
Padrões persistentes e desproporcionais sem explicação clínica (ex.: LR altos por longos períodos, fora de infecções/vacinas)
Nesses cenários, o caminho é discutir com o pediatra, considerar repetição do exame, revisão do esfregaço e, se indicado, investigação direcionada.
Erros comuns (e como evitá-los)
Nunca “liberar” LR em crianças
Errado. Em crianças pequenas, LR são comuns. Omiti-los prejudica a leitura do caso.
Só “liberar” LR quando há leucocitose
Errado. LR podem aparecer com leucócitos normais. O achado é morfológico/funcional, não depende da contagem total.
Laudo genérico, sem contexto
Melhor: incluir observações curtas e úteis (“achado compatível com faixa etária/reatividade imunológica”, “correlacionar com quadro clínico”).
Boas práticas para laboratórios e profissionais
Padronize critérios de identificação e relato de LR (checklists morfológicos ajudam).
Comunique “sem drama”: use linguagem clara e educativa no comentário do laudo.
Integre com a clínica: se houver sinal de alerta, destaque e sugira correlação/seguimento.
Eduque pais/cuidadores: quando houver contato direto, explique que LR é, na maioria, “sinal de defesa”, não de gravidade.
Exemplo de comentário de laudo (sugestão):
“Presença de linfócitos reativos, achado comum na faixa etária pediátrica e compatível com resposta imune. Correlacionar com quadro clínico. Sem outras atipias relevantes no esfregaço.”
Para pais e cuidadores: uma explicação simples
“Linfócito reativo” significa que células de defesa do seu filho foram ativadas — geralmente por contato com vírus, bactérias, vacinas ou outros estímulos normais nesta fase da vida. Na grande maioria dos casos, não é motivo de pânico. O médico avalia o resultado junto com os sintomas e com o restante do exame.
Perguntas frequentes (FAQ)
1) LR é sinal de infecção grave?
Na maioria das vezes, não. É um marcador de resposta imune, comum em resfriados, pós-vacina e outras exposições benignas.
2) Precisa repetir o hemograma por causa de LR?
Depende do contexto. Se a criança está bem e o restante do hemograma está ok, geralmente não.
3) LR vira “câncer de sangue”?
LR não são blastos. São células maduras ativadas. Preocupações com hematopatias surgem por conjunto de achados (morfologia suspeita, citopenias, clínica), não por LR isolado.
4) Existe “valor normal” de LR?
Relata-se presença e percentual; não há um “valor de referência” universal — o importante é a interpretação contextual (idade, clínica, demais parâmetros).
Principais aprendizados
Em crianças (RN → ~4 anos), linfócito reativo = achado esperado.
Não condicione o relato de LR à presença de leucocitose.
Descreva e contextualize: ajuda o pediatra e evita alarmes desnecessários.
Fique atento aos sinais de alerta para diferenciar reatividade fisiológica de situações que pedem investigação.
No HemoClass LAN você entenderá DEFINITIVAMENTE as alterações hematológicas com base na fisiopatologia de cada alteração/processo.
O LAN é disparado a MELHOR FORMAÇÃO em hematologia do mercado!
png.jpg)
Howell Jolly Like em Neutrófilos
Warning: strtotime(): It is not safe to rely on the system's timezone settings. You are *required* to use the date.timezone setting or the date_default_timezone_set() function. In case you used any of those methods and you are still getting this warning, you most likely misspelled the timezone identifier. We selected the timezone 'UTC' for now, but please set date.timezone to select your timezone. in /home/hemocla/public_html/site/blog.php on line 179
Warning: date(): It is not safe to rely on the system's timezone settings. You are *required* to use the date.timezone setting or the date_default_timezone_set() function. In case you used any of those methods and you are still getting this warning, you most likely misspelled the timezone identifier. We selected the timezone 'UTC' for now, but please set date.timezone to select your timezone. in /home/hemocla/public_html/site/blog.php on line 179
13/10/2025 |
#Leucemia
#Leucograma
#Hematoscopia
1) O que são
Inclusões citoplasmáticas arredondadas, pequenas, fortemente basofílicas (roxo‑escuro) em neutrófilos, que lembram visualmente os corpos de Howell‑Jolly dos eritrócitos. Estudos com coloração fluorescente (p.ex., DAPI) confirmam natureza DNA‑positiva (fragmentos nucleares).
Importante: não confundir com corpos de Howell‑Jolly em hemácias (marcadores de hipo/asplenia). As HJLI ocorrem em neutrófilos e estão ligadas principalmente a imunossupressão e granulopoiese estressada/displásica.
2) Características morfológicas (como reconhecer)
Formato/contorno: esférico a oval, bem delimitado.
Tamanho: pequeno (tipicamente < grânulo tóxico; semelhante ou um pouco maior que um ponto de cromatina).
Cor: basofílica intensa em colorações tipo Romanowsky (roxo escuro).
Número/posição: geralmente única por célula, localizada no citoplasma; pode haver raras múltiplas.
Células acometidas: predominantemente neutrófilos segmentados e bastonetes.
Dicas práticas
Aumente para imersão (100×) para avaliar contorno nítido e cor homogênea.
Se disponível, contraste com Döhle (azul‑claro mal delimitado) e com mórulas de Anaplasma/Ehrlichia (aglomerados múltiplos, maiores, aspecto “saco de uvas”).
3) Diagnóstico diferencial rápido
Achado
Localização
Cor/aspecto
Dicas diferenciais
HJLI (neutrófilo)
Citoplasma
Ponto roxo‑escuro, redondo, bem nítido
Geralmente único; DNA‑positivo; associação com imunossupressão/terapia
Döhle
Citoplasma
Mancha azul‑pálida, mal definida
ER rugoso; costuma vir com toxicidade (grânulos tóxicos, vacúolos)
Mórula (Anaplasma/Ehrlichia)
Citoplasma
Aglomerado de grânulos finos
Várias “bolinhas” juntas; quadro febril/leucopenia plausível
Corpúsculo de Barr
Núcleo (apêndice)
“Drumstick” ligado à cromatina
Visto em mulheres; é apêndice nuclear, não inclusão citoplasmática
4) Como reportar no laudo
Campo: Morfologia leucocitária / observações
Modelo 1 (achado discreto): “Inclusões tipo Howell‑Jolly em neutrófilos, raras.”
Modelo 2 (achado significativo): “Inclusões tipo Howell‑Jolly em neutrófilos: ocasionais a frequentes. Achado descrito em pacientes imunossuprimidos (p.ex., pós‑transplante, HIV, quimioterapia) e em granulopoiese displásica; correlacionar com quadro clínico/terapêutico.”
Quantificação sugerida (opcional): classificar como raras (<1/100 neutrófilos), ocasionais (1–5/100), frequentes (>5/100), conforme contagem estimada em 200 leucócitos.
Quando acrescentar comentário: se novo no prontuário, se numerosas, ou se não há histórico de imunossupressão conhecido.
Exemplo de comentário curto:
“As inclusões observadas têm morfologia compatível com Howell‑Jolly‑like em neutrófilos, descritas em contextos de imunossupressão (p.ex., antivirais análogos de nucleosídeos, tacrolimo, quimioterapia), infecções virais e displasia granulocítica. Sugerimos correlação com medicações, histórico de transplante/HIV e achados clínico‑laboratoriais associados.”
5) Significado clínico e associações descritas
Imunossupressão: pós‑transplante sólido ou de células‑tronco; uso de tacrolimo/calcineurínicos, quimioterapia.
Antivirais (análogos de nucleosídeos) e terapia antirretroviral.
HIV/AIDS e infecções virais (ex.: relatos em COVID‑19).
Síndromes mielodisplásicas (SMD) e granulopoiese estressada/displásica.
Interpretação: achado inespecífico, porém sinalizador de contexto clínico. Não confere, por si, diagnóstico etiológico ou prognóstico independente; deve ser interpretado junto a história, medicações e demais alterações morfológicas (p.ex., pseudo‑Pelger, grânulos tóxicos).
6) Recomendações operacionais para o laboratório
Confirmar em imersão e por dupla leitura quando possível.
Registrar e graduar (rara/ocasional/frequente) no LIS.
Verificar histórico no pedido/prontuário: transplante, HIV, quimioterapia, antivirais/imunossupressores.
Sugerir correlação clínica no laudo quando achado for novo ou proeminente.
Em suspeita clínica de anaplasmose/ehrlichiose (febre, citopenias, exposição a carrapato), alertar o médico e considerar testes específicos.
7) Ilustrações (para treinamento interno)
Exemplos de HJLI em neutrófilos (banco de imagens hematológicas em materiais didáticos do serviço).
Painel comparativo com Döhle, mórulas e corpúsculo de Barr.
8) Nota de educação continuada
Este informativo sintetiza evidências de relatos de caso, séries e revisões recentes. O fenômeno é raro, porém relevante em populações imunossuprimidas. Manter a equipe treinada para não confundir com inclusões infecciosas ou alterações nucleares fisiológicas (Barr).

Alterações Hematológicas no Uso Crônico de Álcool: Causas, Diagnóstico e Apoio Laboratorial
Warning: strtotime(): It is not safe to rely on the system's timezone settings. You are *required* to use the date.timezone setting or the date_default_timezone_set() function. In case you used any of those methods and you are still getting this warning, you most likely misspelled the timezone identifier. We selected the timezone 'UTC' for now, but please set date.timezone to select your timezone. in /home/hemocla/public_html/site/blog.php on line 179
Warning: date(): It is not safe to rely on the system's timezone settings. You are *required* to use the date.timezone setting or the date_default_timezone_set() function. In case you used any of those methods and you are still getting this warning, you most likely misspelled the timezone identifier. We selected the timezone 'UTC' for now, but please set date.timezone to select your timezone. in /home/hemocla/public_html/site/blog.php on line 179
11/08/2025 |
#Padronização
#Sindromes
#exames
Alterações Hematológicas no Uso Crônico de Álcool: Como Identificar e Interpretar no Laboratório
Na rotina de um laboratório clínico, não é incomum receber hemogramas com alterações que, à primeira vista, parecem não ter explicação. Trombocitopenia repentina, pancitopenia sem causa aparente, anemias incomuns — tudo isso pode levantar dúvidas e atrasar a liberação de resultados.
Em muitos desses casos, um fator comum aparece quando investigamos mais a fundo: o uso crônico de álcool.
Por que o uso crônico de álcool altera o hemograma?
O etanol exerce múltiplos efeitos nocivos sobre o sistema hematológico, e compreender esses mecanismos é essencial para evitar erros interpretativos:
Toxicidade medular: o álcool afeta diretamente a medula óssea, reduzindo a produção de eritrócitos, leucócitos e plaquetas, levando à pancitopenia periférica.
Deficiências nutricionais: compromete a absorção de vitaminas essenciais à hematopoese. A falta de vitamina B12 e ácido fólico causa anemia megaloblástica, enquanto a deficiência de vitamina B6 leva à anemia sideroblástica.
Síndrome de Zieve: caracterizada por anemia hemolítica, icterícia e hiperlipidemia, geralmente associada à deficiência de vitamina E e alterações na membrana eritrocitária.
Trombocitopenia alcoólica: pode ocorrer pela redução da trombopoietina hepática ou pelo hiperesplenismo decorrente de hipertensão portal em hepatopatias alcoólicas.
Apoptose plaquetária: o álcool pode induzir morte celular precoce nas plaquetas, aumentando o risco de sangramento.
Imunidade comprometida: o uso crônico prejudica a função de neutrófilos e macrófagos, elevando a suscetibilidade a infecções.
A importância da interpretação correta no laboratório
Nem sempre o prontuário do paciente traz informações claras sobre histórico de etilismo. Muitas vezes, o analista clínico precisa suspeitar do uso crônico de álcool a partir dos padrões hematológicos observados e buscar exames complementares para confirmar.
Uma interpretação segura depende de:
Correlação entre achados hematológicos e bioquímicos;
Conhecimento atualizado sobre mecanismos fisiopatológicos;
Atenção aos sinais indiretos no hemograma.
Quando buscar apoio especializado?
Casos complexos exigem decisões rápidas e embasadas. Ter acesso a uma assessoria remota em hematologia significa contar com suporte técnico imediato para discutir resultados, orientar investigações adicionais e padronizar laudos.
Esse suporte é especialmente valioso quando:
O hemograma apresenta alterações múltiplas e atípicas;
Há suspeita de condições como Síndrome de Zieve ou anemia sideroblástica;
O histórico clínico é incompleto ou inexistente.
Com esse tipo de parceria, o laboratório aumenta sua assertividade, reduz erros interpretativos e entrega mais valor para médicos e pacientes.
📌 Sua equipe pode interpretar hemogramas complexos com muito mais segurança. Conheça como nossa assessoria remota pode integrar conhecimento técnico-laboratorial à sua rotina e elevar a qualidade dos seus resultados.
Nos contate clicando AQUI para saber mais sobre o serviço de assessoria remota!
Texto produzido por Flavio Simplicio Maia - Assessor HemoClass

Leucemia ou Linfoma de Burkitt: Entenda a Atualização na Classificação
Warning: strtotime(): It is not safe to rely on the system's timezone settings. You are *required* to use the date.timezone setting or the date_default_timezone_set() function. In case you used any of those methods and you are still getting this warning, you most likely misspelled the timezone identifier. We selected the timezone 'UTC' for now, but please set date.timezone to select your timezone. in /home/hemocla/public_html/site/blog.php on line 179
Warning: date(): It is not safe to rely on the system's timezone settings. You are *required* to use the date.timezone setting or the date_default_timezone_set() function. In case you used any of those methods and you are still getting this warning, you most likely misspelled the timezone identifier. We selected the timezone 'UTC' for now, but please set date.timezone to select your timezone. in /home/hemocla/public_html/site/blog.php on line 179
06/03/2025 |
A classificação das leucemias e linfomas evoluiu ao longo dos anos, trazendo mudanças significativas na forma como algumas doenças são categorizadas. Um dos exemplos mais marcantes é a leucemia de Burkitt, que antes era classificada como LLA-L3 na antiga classificação FAB (French-American-British). No entanto, de acordo com as diretrizes mais recentes da Organização Mundial da Saúde (OMS), essa denominação não é mais utilizada.
A Classificação FAB e a LLA-L3
Antigamente, a classificação FAB dividia as leucemias linfoblásticas agudas (LLA) em três subtipos principais, com base nas características morfológicas das células:
LLA-L1: Blastos pequenos e homogêneos.
LLA-L2: Blastos grandes e heterogêneos.
LLA-L3: Blastos grandes com citoplasma vacuolizado, característicos da leucemia de Burkitt.
Essa categorização se baseava exclusivamente na morfologia celular observada no esfregaço sanguíneo ou na medula óssea. Assim, as células da leucemia de Burkitt eram chamadas de blastos e encaixadas na LLA-L3.
A Nova Classificação da OMS: O Que Mudou?
Com os avanços na imunofenotipagem, citogenética e biologia molecular, descobriu-se que as células da leucemia de Burkitt não são realmente blastos imaturos, como ocorre nas demais LLAs, mas sim linfócitos B maduros anômalos.
Por isso, a classificação da OMS atualizou o conceito:
🔬 A leucemia de Burkitt não é mais considerada uma LLA, mas sim um linfoma de células B maduras com envolvimento leucêmico.
Isso significa que:
✅ As células que antes eram chamadas de blastos LLA-L3 agora são reconhecidas como linfócitos B maduros.
✅ A doença passou a ser classificada dentro do grupo dos linfomas de células B maduras.
✅ Quando há envolvimento da medula óssea ≥ 20%, ainda se usa o termo leucemia de Burkitt, mas o conceito baseia-se na origem celular madura e não em um blasto precursor.
Por Que Essa Mudança é Importante?
A atualização da OMS reflete um entendimento mais preciso da biologia da doença. A leucemia de Burkitt:
📌 Origina-se de células B maduras e não de blastos imaturos, como nas LLAs clássicas.
📌 Apresenta rearranjos no gene MYC, um marcador genético característico.
📌 Possui um comportamento clínico agressivo, típico dos linfomas de alto grau.
Essa mudança afeta diretamente a forma como os profissionais interpretam os exames laboratoriais e direcionam o tratamento.
Conclusão: A Leucemia de Burkitt e a Prática Laboratorial
Para os profissionais que trabalham na análise de leucemias, essa mudança reforça a importância de não se basear apenas na morfologia celular para a classificação das doenças hematológicas. A imunofenotipagem e a biologia molecular são ferramentas fundamentais para um diagnóstico preciso e atualizado.
Se você quer entender mais sobre a classificação das leucemias, interpretação laboratorial e diagnóstico diferencial, conheça o Curso Hemoclass Leucemias. Lá, você terá acesso a conteúdos aprofundados, baseados nas diretrizes mais atuais, para aprimorar suas análises laboratoriais com segurança e embasamento técnico.
🔗 Clique AQUI e saiba mais sobre o Hemoclass Leucemias!
link direto:
https://hemoclasscursos.com.br/hemoclass-leucemias/

Contagem Manual de Plaquetas: Veja Quando e Como Usar.
Warning: strtotime(): It is not safe to rely on the system's timezone settings. You are *required* to use the date.timezone setting or the date_default_timezone_set() function. In case you used any of those methods and you are still getting this warning, you most likely misspelled the timezone identifier. We selected the timezone 'UTC' for now, but please set date.timezone to select your timezone. in /home/hemocla/public_html/site/blog.php on line 179
Warning: date(): It is not safe to rely on the system's timezone settings. You are *required* to use the date.timezone setting or the date_default_timezone_set() function. In case you used any of those methods and you are still getting this warning, you most likely misspelled the timezone identifier. We selected the timezone 'UTC' for now, but please set date.timezone to select your timezone. in /home/hemocla/public_html/site/blog.php on line 179
23/02/2025 |
#Hemostasia e Coagulação
#Coagulograma
Principais Métodos de Contagem Manual de Plaquetas
Se tem uma coisa que um bom analista clínico precisa entender, é que a contagem automatizada de plaquetas é sempre a melhor escolha Além de ser mais prática, ela entrega resultados com muito mais precisão. Mas, como nem tudo são flores no laboratório, existem situações em que a contagem manual se faz necessária – seja para confirmar um resultado duvidoso ou quando o equipamento simplesmente se recusa a colaborar.
🔬 1. Método de Fônio
Este método utiliza aumento de 100X e compara a quantidade de plaquetas com 1.000 hemácias. Depois, basta multiplicar pelo número total de hemácias por mL.
📌 Exemplo:
- Foram contadas 35 plaquetas em 10 campos, com 4,2 milhões de hemácias.
- Resultado final:147.000 plaquetas
---
🔬 2. Método de Bárbara H. O’Connor
Aqui, a contagem ocorre em 10 campos no aumento de 100X. Depois, faz-se uma média e multiplica-se por 13.000.
📌 Exemplo:
- A média da contagem nos 10 campos foi de 14 plaquetas.
- Resultado final: 182.000 plaquetas
---
🔬 3. Método Nosanchuk, Chang e Bennett
Este método conta plaquetas em **8 campos** no aumento de 100X e, depois, multiplica-se o total obtido por 2.000.
📌 Exemplo:
- Contagem total nos 8 campos: 85 plaquetas.
- Resultado final: 170.000 plaquetas
---
🔬 4. Método Alternativo (Apenas para Confirmação)
Essa técnica faz uma média das plaquetas em 10 campos, multiplica pelo valor da hemoglobina e depois por 1.000.
📌 Exemplo:
- Média de 14 plaquetas por campo e hemoglobina de 12 g/dL.
- Resultado final: 168.000 plaquetas
---
🔬 5. Método de Rees-Ecker (Câmara de Neubauer)
Utiliza aumento de 40X e consiste na diluição da amostra, que é inserida na câmara de Neubauer. A contagem ocorre no quadrante central e o valor obtido é multiplicado por 1.000.
---
🚨 Afinal, a Contagem Manual é Confiável?
Agora vem a grande questão: podemos confiar nas contagens manuais?
A resposta curta é não totalmente. Todos os métodos manuais apresentam baixa exatidão e não devem substituir a contagem automatizada.
O que fazer então?
✔ Mantenha seu equipamento sempre calibrado e com manutenção em dia.
✔ Faça o controle de qualidade regularmente.
✔ Confie no seu analisador hematológico!
Se houver necessidade de um método manual, o de Rees-Ecker na câmara de Neubauer ainda é o mais confiável. Mas lembre-se: a contagem manual é um plano B, nunca o plano A!
🔎 E você, já teve que recorrer à contagem manual? Qual é o método padronizado no seu laboratório? Se não tem nenhum, é preciso pensar nisso!
---
Agora, um passo além: Você realmente entende a avaliação das plaquetas?
Saber contar plaquetas é só o começo! A verdadeira análise vem com a interpretação correta dos parâmetros plaquetários e do coagulograma.
Se você quer finalmente dominar essa etapa essencial da hematologia, entender os novos parâmetros plaquetários e saber como interpretar cada detalhe sem erros, precisa conhecer o nosso eBook exclusivo sobre o tema!
📥 Conheça agora o eBook sobre Interpretação do Coagulograma e Novos Parâmetros Plaquetários e transforme sua forma de avaliar hemogramas!
Clique AQUI para saber mais sobre esse ebook!
Se não der certo, copie e cole o link abaixo no seu navegador:
https://hemoclasscursos.com.br/ic/

Bastonete de Auer: O Achado que Você Não Pode Ignorar no Hemograma!
Warning: strtotime(): It is not safe to rely on the system's timezone settings. You are *required* to use the date.timezone setting or the date_default_timezone_set() function. In case you used any of those methods and you are still getting this warning, you most likely misspelled the timezone identifier. We selected the timezone 'UTC' for now, but please set date.timezone to select your timezone. in /home/hemocla/public_html/site/blog.php on line 179
Warning: date(): It is not safe to rely on the system's timezone settings. You are *required* to use the date.timezone setting or the date_default_timezone_set() function. In case you used any of those methods and you are still getting this warning, you most likely misspelled the timezone identifier. We selected the timezone 'UTC' for now, but please set date.timezone to select your timezone. in /home/hemocla/public_html/site/blog.php on line 179
18/02/2025 |
#Leucemia
#Sindromes
#HemoClass Leucemias
O Que São os Bastonetes de Auer?
São estruturas citoplasmáticas que lembram pequenas agulhas alongadas, de coloração rosa-avermelhada. São compostos por enzimas lisossomais, peroxidase e inclusões cristalinas – um verdadeiro coquetel químico dentro da célula.
Mas aqui vai um detalhe que muita gente esquece: apesar de serem um forte indicativo de LMA, os bastonetes de Auer também podem aparecer em algumas síndromes mielodisplásicas. Ou seja, se você encontrou um, fique atento!
Como Relatar no Hemograma?
Se o blasto tem bastonete de Auer, ele deve ser contado como blasto na contagem diferencial e descrito cuidadosamente nas observações do hemograma. Nada de soltar essa informação de forma genérica! O ideal é relatar:
✅ A presença do bastonete de Auer
✅ Outras características morfológicas do blasto
Por quê? Porque um hemograma que já indica a presença de bastonetes de Auer antecipa o diagnóstico e acelera o atendimento do paciente, identificando desde cedo que a linhagem acometida é mieloide. Isso significa menos tempo perdido e mais chances de intervenção rápida.
Como Identificar Corretamente?
🔬 Aumento de 100X no microscópio
🩸 Corante May-Grunwald-Giemsa (nossa escolha de referência)
🎯 Região correta da lâmina – porque avaliar a área errada pode te enganar feio!
Agora que você já sabe a importância dos bastonetes de Auer, não deixe passar despercebido. Treine seu olhar, revise suas lâminas e veja nas imagens abaixo exemplos clássicos desse achado essencial!





